
Research Interests
Cancer is a disease of defective DNA repair; cancer at its origin must acquire permanent genomic alternations and thus genome instability is one of the key hallmarks of cancer development and progression. Many hematological malignancies and solid tumors arise as a consequence of defects in DNA repair and chromosome aberrations. DNA damage response and DNA repair system constitutes a critical tumor suppressor network to preserve the integrity of the genome, thereby preventing the onset and progression of tumor. Therefore, understanding the basic mechanisms of DNA repair and their deregulation in specific types of cancer allows us to better appreciate tumorigenesis and come up with personalized strategies for treating cancer patients.
1. Genome stability at replication forks - Replication stress response
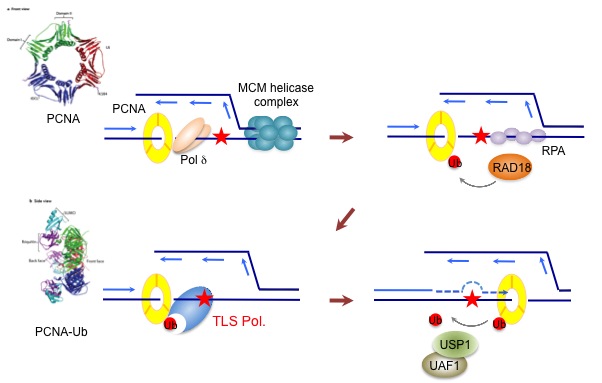 Figure 1. DNA replication stress response pathway
Figure 1. DNA replication stress response pathway
Achieving accurate genome duplication is a challenging task for the DNA replication machinery. Failure to overcome various replication-blocking obstacles causes perturbations of DNA synthesis, collectively referred to as DNA replication stress. Increased formation of aberrant replication fork structures due to the loss of replication stress response pathways leads to deleterious mutational events that are highly associated with genome instability and tumorigenesis. As such, elevated DNA replication stress is prevalent in cancer. However, this also provides a therapeutic opportunity where we can exacerbate the replication stress of cancer cells to achieve specific killing of cancer cells, as exemplified by a recent development of checkpoint inhibitors for cancer therapy.
The key to successful DNA replication is the ability of cells to deploy a battery of genome maintenance proteins that preserve replisome integrity, protect and restart stalled replication forks. Poly(ADP-ribose); PAR polymerase 1 (PARP1) is a multifaceted protein involved in various DNA repair processes by modifying target proteins and itself with PAR chains. Emerging evidence supports that PARP1 activity regulates replication fork progression and the stability of stalled forks, highlighting the specialized role of PARP1 at the DNA replication fork. Consequently, PARP1 inhibition is being exploited clinically to induce synthetic lethality with BRCA deficiency by exacerbating the instability of stalled replication forks. Nevertheless, how PARP1 activity is connected to the DNA replication machinery remains largely elusive. Understanding its concerted actions that occur under the control of replisome activity should provide an important clue for the genome maintenance processes at the replication fork. We are interested in understanding the roles of TIMELESS (TIM)-TIPIN in the fork protection complex (FPC), an obligate heterodimer of the replisome, in preserving DNA replication fork integrity via its interplay with multiple genome surveillance proteins at DNA replication forks, including PARP1. The FPC acts as a scaffold that tethers the helicase-polymerase movement to prevent uncoupling of their activities, which would otherwise result in disintegration of the replisome and extensive single-stranded DNA (ssDNA) formation. Loss of the FPC leads to DNA replication defect and fork stalling, indicating that maintaining structural integrity of the replisome is critical for preserving fork stability. Recently, we demonstrated that that TIM is regulated by its interaction with SDE2, a replication stress regulator that we first identified its role in DNA replication under damage, and elucidated its roles engaged at both active and stalled forks. We are currently investigating mechanisms by which the FPC protects DNA replication fork integrity and how its deregulation is connected to tumorigenesis. Fundamental knowledge on DNA replication and stress responses will provide opportunities to exploit the replication vulnerabilities of cancer cells for therapy (Supported by NIGMS R01 and American Cancer Society).
2. The Fanconi anemia (FA) DNA interstrand cross-link repair pathways
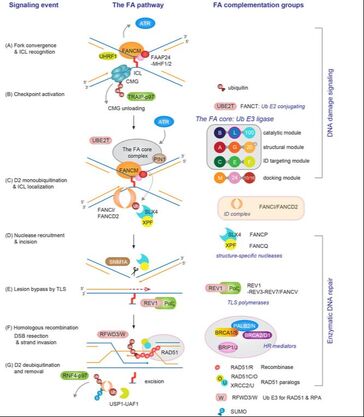 Figure 2. The FA pathway
Figure 2. The FA pathway
The FA pathway resolves stalled replication forks in response to DNA interstrand cross-links (ICLs), lethal lesions that block DNA replication and transcription. Currently, 22 FA gene products constitute the FA pathway, and germ-line mutations in this pathway lead to bone marrow failure and cancer predisposition. Central to the FA pathway is monoubiquitination of the FANCD2 protein by the FA core ubiquitin E3 ligase complex, which in turn recruits enzymes necessary for processing ICLs. Thus, the activity of the FA core complex needs to be tightly controlled to recognize lesions and initiate repair. In addition to its catalytic core, the FA core complex is composed of multiple subunits that can be potentially regulated by a variety of posttranslational modifications. We are focusing on the proteolytic signaling that control the activity of the FA core complex, which would dictate the outcome of DNA ICL repair and responses to cytotoxic chemotherapy, and investigating the impact of its deregulation to tumorigenesis (Supported by NCI R01).
